量子化学模拟计算在解析微观尺度上表面催化反应及材料特性等领域中发挥重要作用。近日,我院李红娇特聘副研究员与天津大学张兵教授、于一夫教授团队合作,负责三步接力催化实现超低过位硝酸盐电还原过程的理论模拟工作,以共同一作在Nature Catalysis上发表相关工作论文。
理论模拟工作部分结果在正文中的表达图
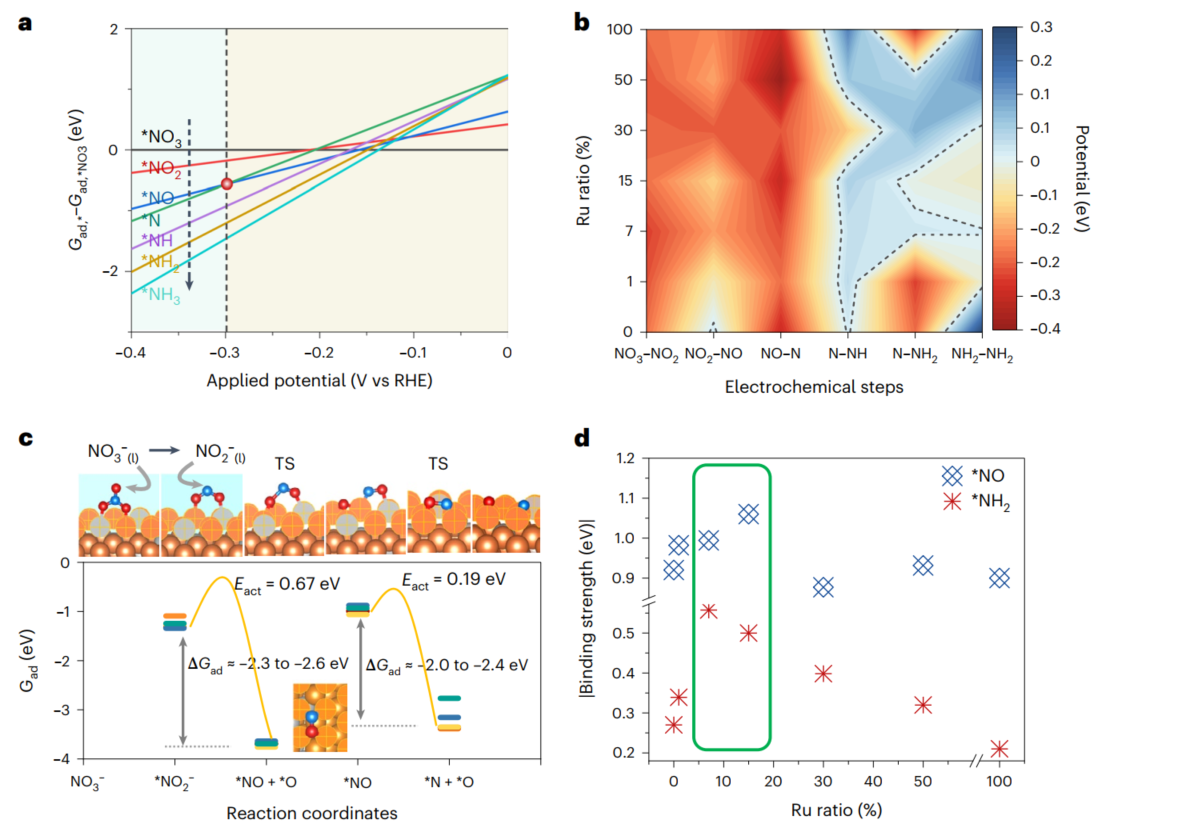
该理论模拟工作主要采用密度泛函理论(DFT)计算解析了RuCo合金催化剂上的硝酸盐电化学合成氨过程。首先,系统的模拟了反应相关物种可能的表面吸附状态,基于此,绘制了RuCo合金催化剂上硝酸根电化学还原反应过程中表面物种Gibbs自由能的热力学相图、表面基元反应热力学驱动力与过电位及合金中元素比例的相图。研究结果推断出硝酸根电还原过程中的过电位控制步骤为*NO到*N的转变,并发现含N-O键断裂过程的基元反应如果经由电化学过程发生,所需过电位远大于实验测试值,从而验证了耦合催化/电催化的三步接力催化过程的必然性。之后,对基元反应相图中存在的控制步骤进行了动力学分析,结果表明该过程存在N-O键断裂的可行化学步骤,过渡态构型指出在合金表面上Ru1Co3四原子团簇为最高活性位。最后,模拟结果发现*NH2和*NO的吸附能随合金比例变化存在火山形曲线,顶点区域对应合金比例与实验中电化学活性最高的合金比例相吻合。

文章全文链接:https://www.nature.com/articles/s41929-023-00951-2
李红娇副研究员 供稿
钮大文 审核
章鹏 编辑
2023年4月25日
